

【文库筛选】CRISPR筛选冲高分?临床转化来助力
01 前言
2022年3月,Takashi Ishio等人在Blood发表“Genome-wide CRISPR screen identifies CDK6 as a therapeutic target in adult T-cell leukemia/lymphoma”,该研究通过全基因组CRISPR敲除筛选发现多个成人T细胞白血病淋巴瘤(Adult T-cell leukemia/lymphoma, ATLL)特异依赖的关键基因,并针对CDK6深入探究,证明TP53失活的ATLL细胞由于CDK2激活而对CDK6抑制剂更加耐受,联合mTOR抑制剂可以协同抑制不同TP53状态的ATLL细胞。该发现为临床ATLL治疗提供了新的靶点和用药策略。
02 背景介绍
03 研究思路
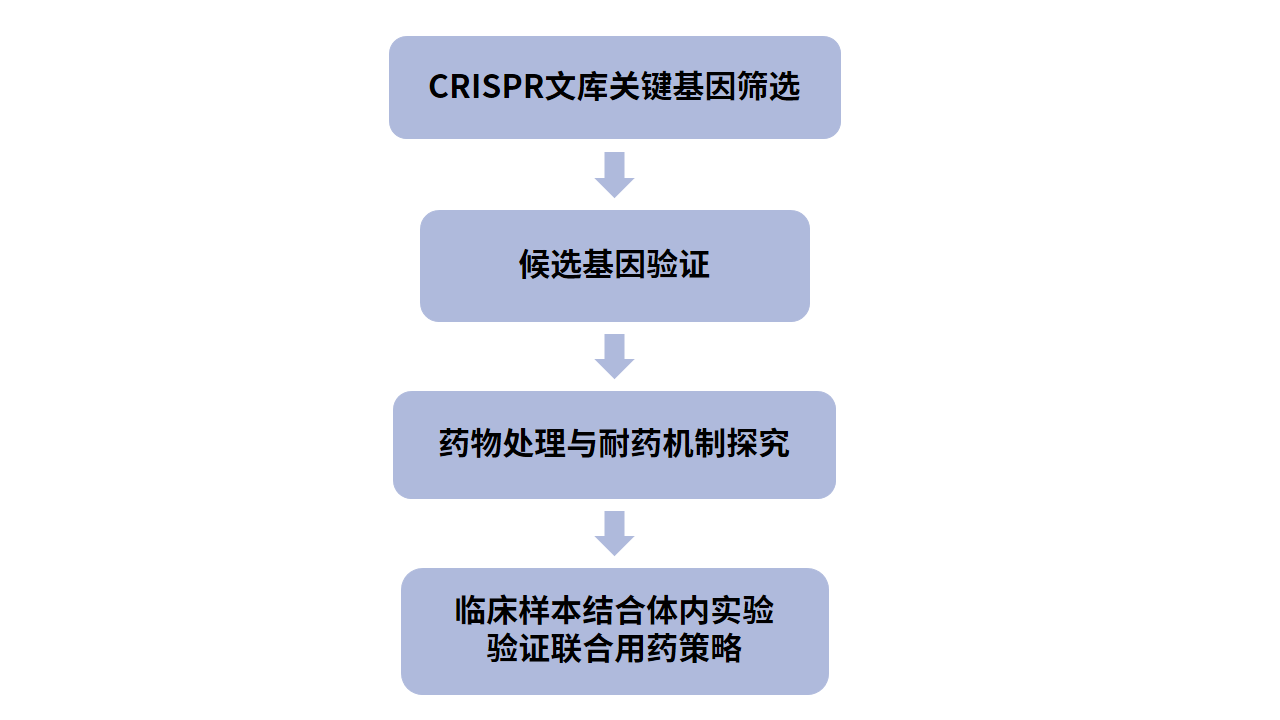
1、通过全基因组CRISPR/Cas9失活筛选寻找ATLL细胞的依赖基因,排除其他细胞普遍依赖的关键基因,确定9个ATLL细胞特异性依赖的候选关键基因。(关键基因筛选)
2、对部分候选基因逐一验证,分别对各候选基因进行敲除,检测对ATLL细胞增殖的影响,然后针对不同候选基因的功能进行相应的实验探究,证明候选基因有临床转化价值。(候选基因验证)
3、以CDK6为切入点,利用临床使用的CDK6抑制剂帕博西尼进行药物处理,证明蛋白水平抑制CDK6能够也能阻滞细胞周期、增加细胞凋亡从而抑制ATLL细胞生长。(抑制剂处理)
4、已知TP53影响CDK6抑制剂治疗效果,并且能够抑制CDK2。通过不同TP53状态的ATLL细胞系,以及构建的TP53敲除细胞系,证明TP53失活的ATLL细胞系对帕博西尼更加耐受。通过进一步敲除CDK2,证明TP53增强帕博西尼对ATLL抑制效果依赖于其对CDK2的抑制。(耐药机制探究)
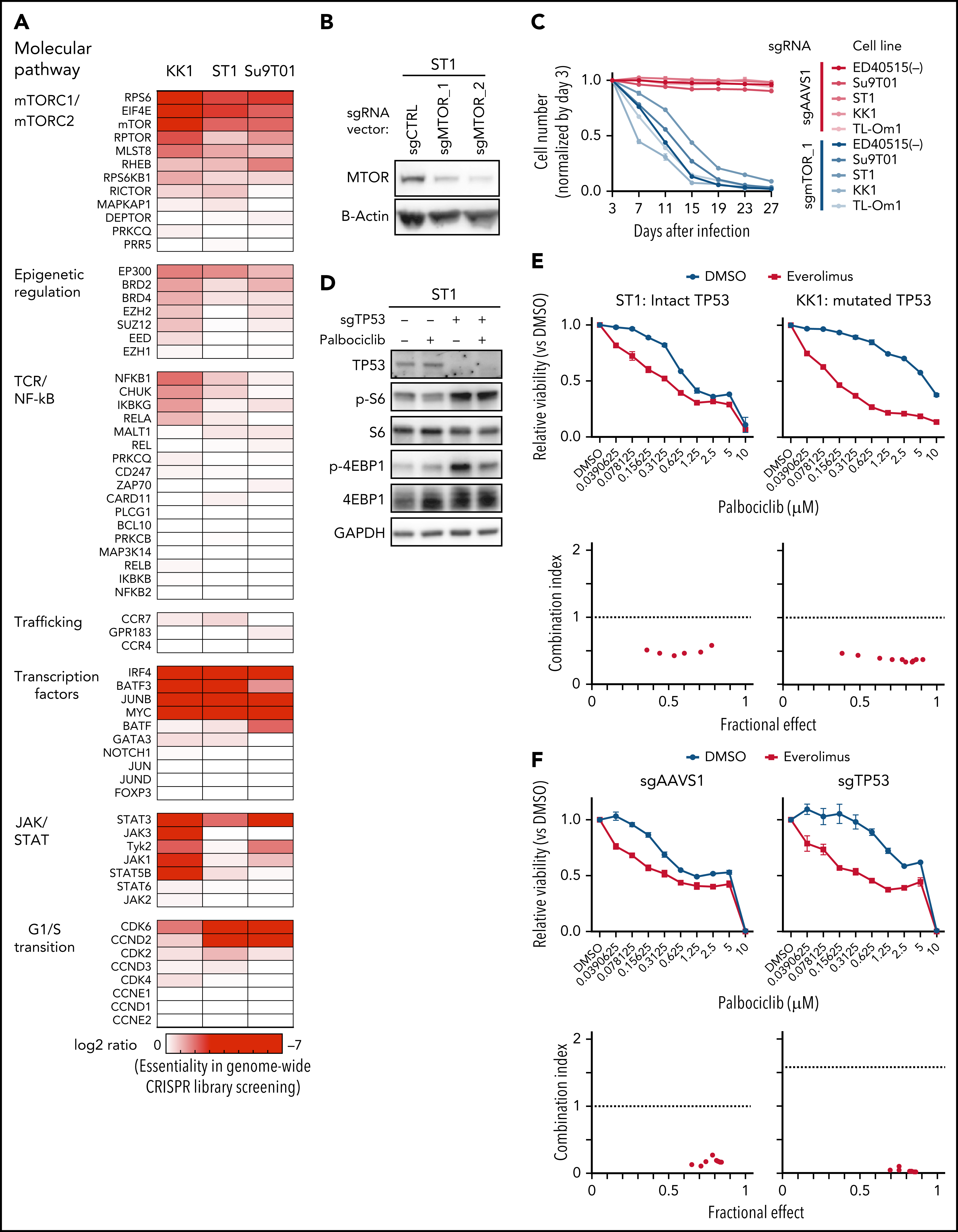
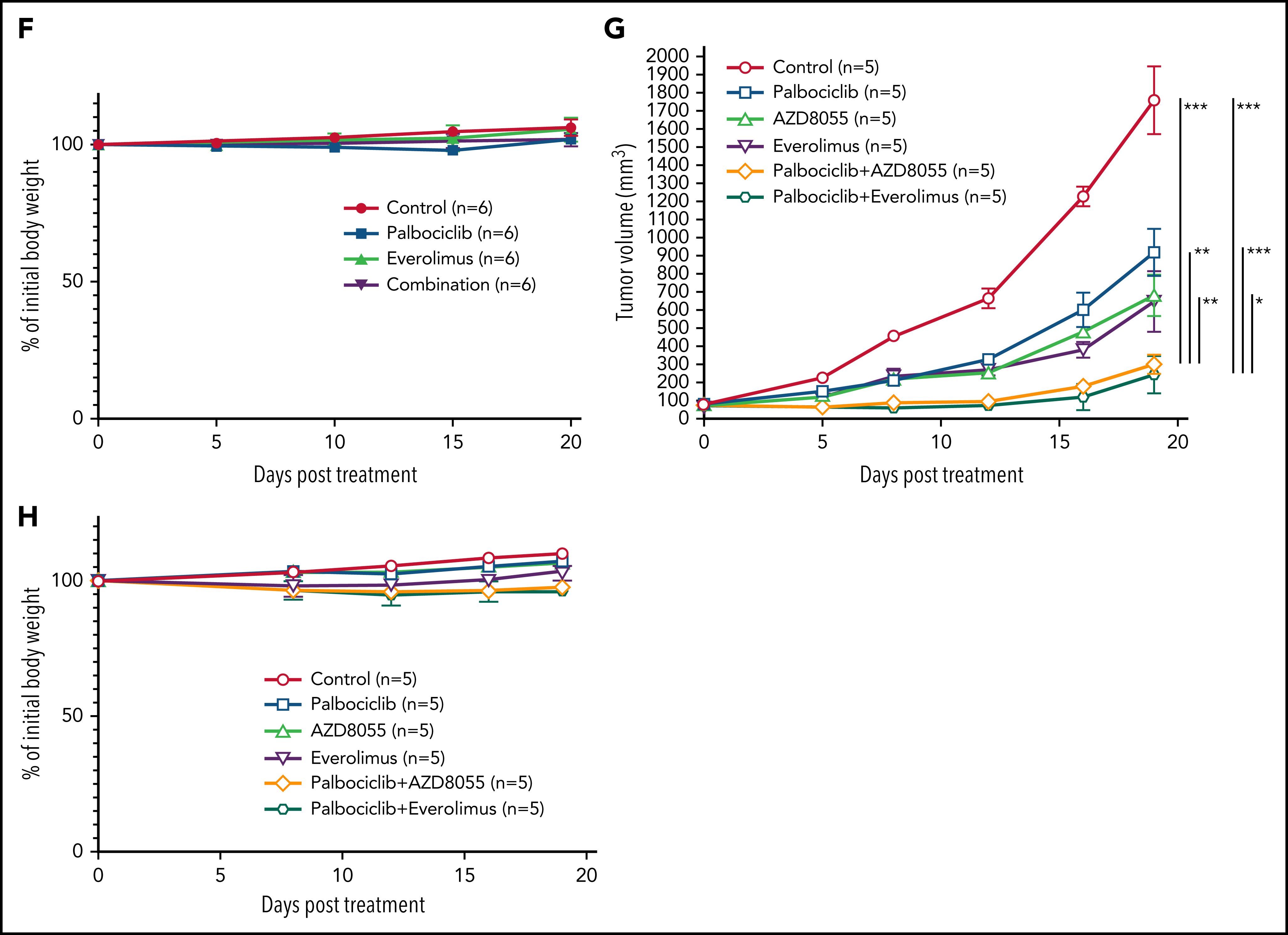
5、CRISPR筛选结合通路分析发现ATLL依赖于mTORC1信号通路,经过细胞增殖以及下游分子磷酸化水平检测的验证后,分别对ATLL细胞系、患者分离的原代细胞以及小鼠异种移植模型进行CDK6和mTOR抑制剂药物处理,在体外和体内水平证明联合用药对任何TP53状态的ATLL治疗效果更加显著。(联合用药策略)
04 研究内容
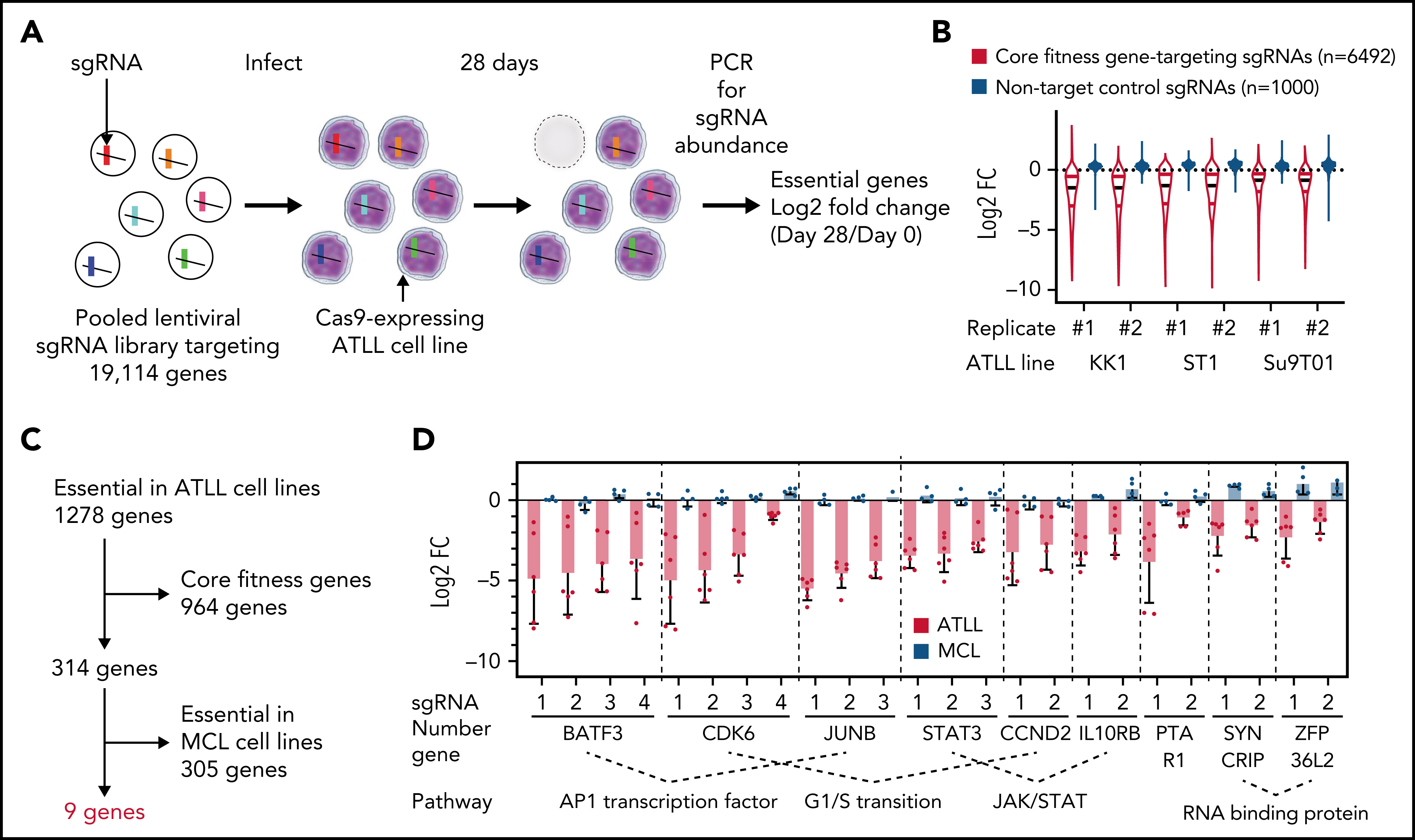
作者对KK1、ST1和Su9T01这3个ATLL细胞系进行全基因组CRISPR失活筛选,在找的1278个关键基因中,排除大多数细胞都依赖的核心关键基因,以及两个套细胞淋巴瘤(mantle cell lymphoma, MCL)细胞系的关键基因,最终确定了9个ATLL细胞特异的候选关键基因。它们分别参与不同的调节通路,包括AP-1家族转录因子(BATF3和JUNB),细胞周期G1-S转换调节因子(CDK6和CCND2),JAK/STAT通路成员(STAT3和IL10RB),RNA结合蛋白(SYNCRIP和ZFP36L2),和一个异戊烯转移酶亚基(PTAR1)。
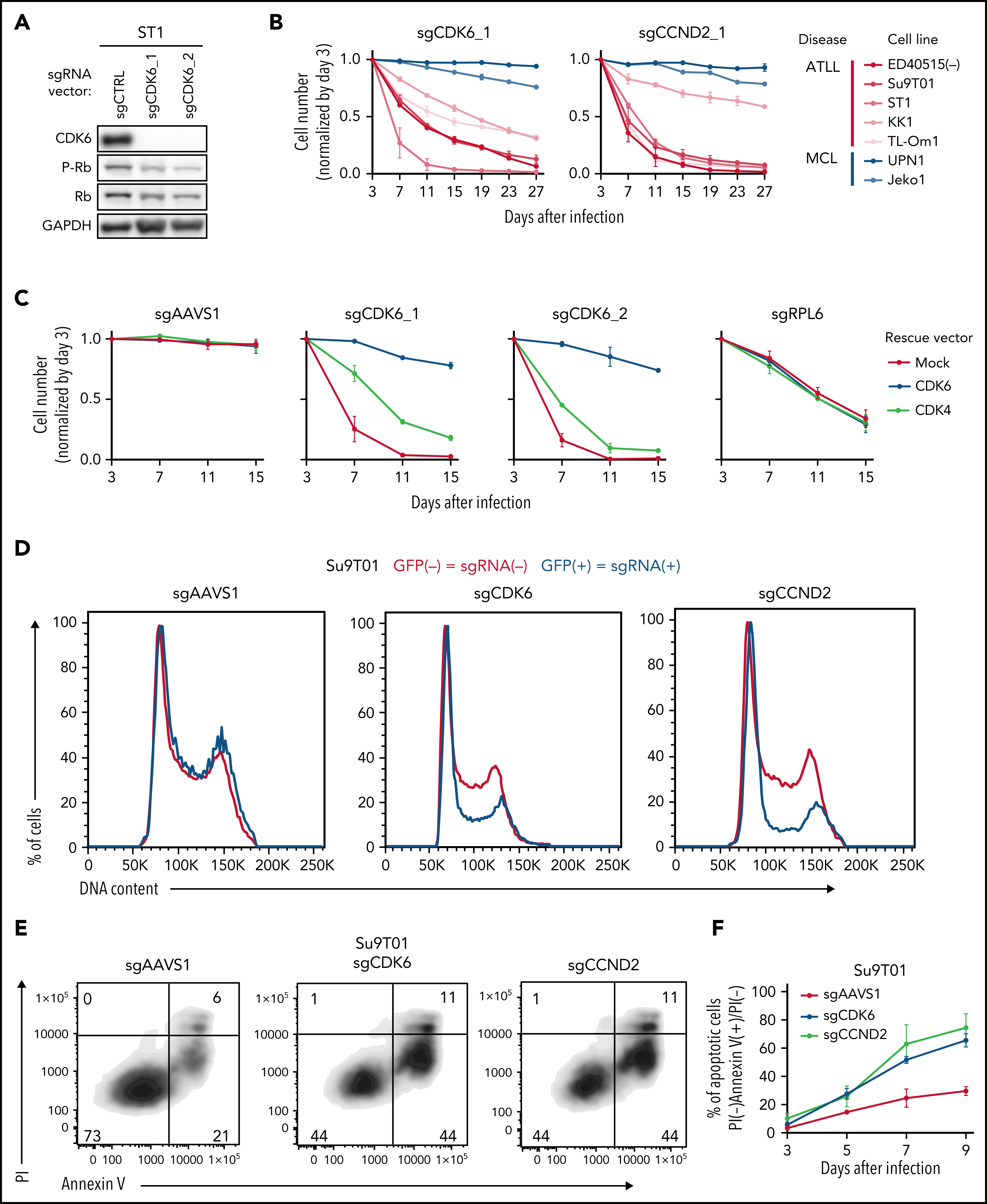
在初步验证部分候选基因后,作者以细胞周期相关候选基因为切入点深入探究。CCND1、CCND2和CCND3是G1/S期的细胞周期蛋白,它们与CDK4或CDK6相互作用形成活性激酶,通过磷酸化并抑制Rb蛋白促进G1-S期转换。免疫印迹显示ATLL细胞敲除CDK6后,Rb磷酸化水平降低。CDK6敲除特异性抑制ATLL细胞,并且可以被CDK6异位表达回补,CDK4异位表达回补效果较弱。与预期一致,CDK6敲除使得ATLL细胞发生G1期阻滞,此外也能诱导细胞凋亡。敲除CCND2与敲除CDK6产生的表型基本一致。
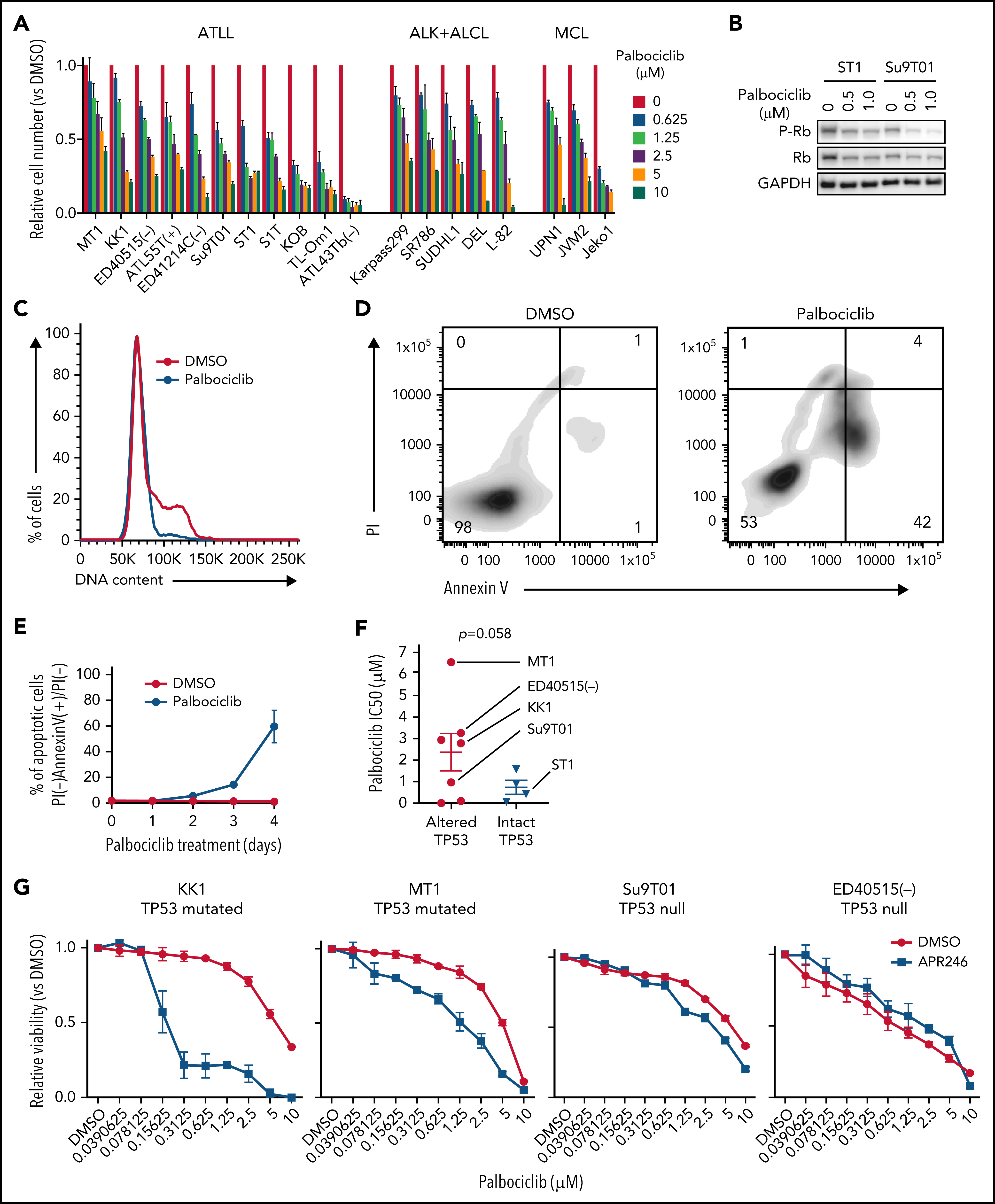
考虑到CDK6对ATLL的抑制作用,作者进一步挖掘CDK6作为ATLL治疗靶点的潜力。研究表明CDK4/6抑制剂帕博西尼能够抑制多种不同细胞系,并且在ATLL细胞系中使得Rb磷酸化水平降低、发生G1期阻滞以及细胞凋亡。既往研究发现在许多不同肿瘤类型中TP53突变与CDK4/6抑制剂耐受相关,将ATLL细胞系按照TP53的状态分组,发现TP53变异(突变或缺失)的ATLL细胞对帕博西尼更加耐受。APR-246是一种能够使突变的TP53恢复转录激活性质的小分子,TP53突变的ATLL细胞经APR-246处理后对帕博西尼更加敏感,但TP53缺失的细胞没有明细的影响。对ST1细胞进行TP53敲除后,在帕博西尼处理下其生长速度高于TP53野生型的ST1细胞。TP53敲除介导的帕博西尼耐受也体现在细胞增殖、周期和凋亡等表型。TP53能通过诱导p21表达阻止G1/S期转换,p21结合并抑制CDK2/Cyclin E。在ST1细胞中,CDK6的失活或抑制会增加p21和CDK2抑制因子p27的表达,但TP53基因失活的ATLL细胞系中不会出现这一情况,表明帕博西尼对ATLL细胞产生理想的抑制效果可能需要抑制CDK2。确实,对TP53突变的KK1和TP53缺失的Su9T01细胞或敲除TP53的ST1细胞在进一步敲除CDK2后,帕博西尼处理下的生长速度下降。至此,作者揭示了TP53能通过抑制CDK2增强ATLL对帕博西尼的敏感性。
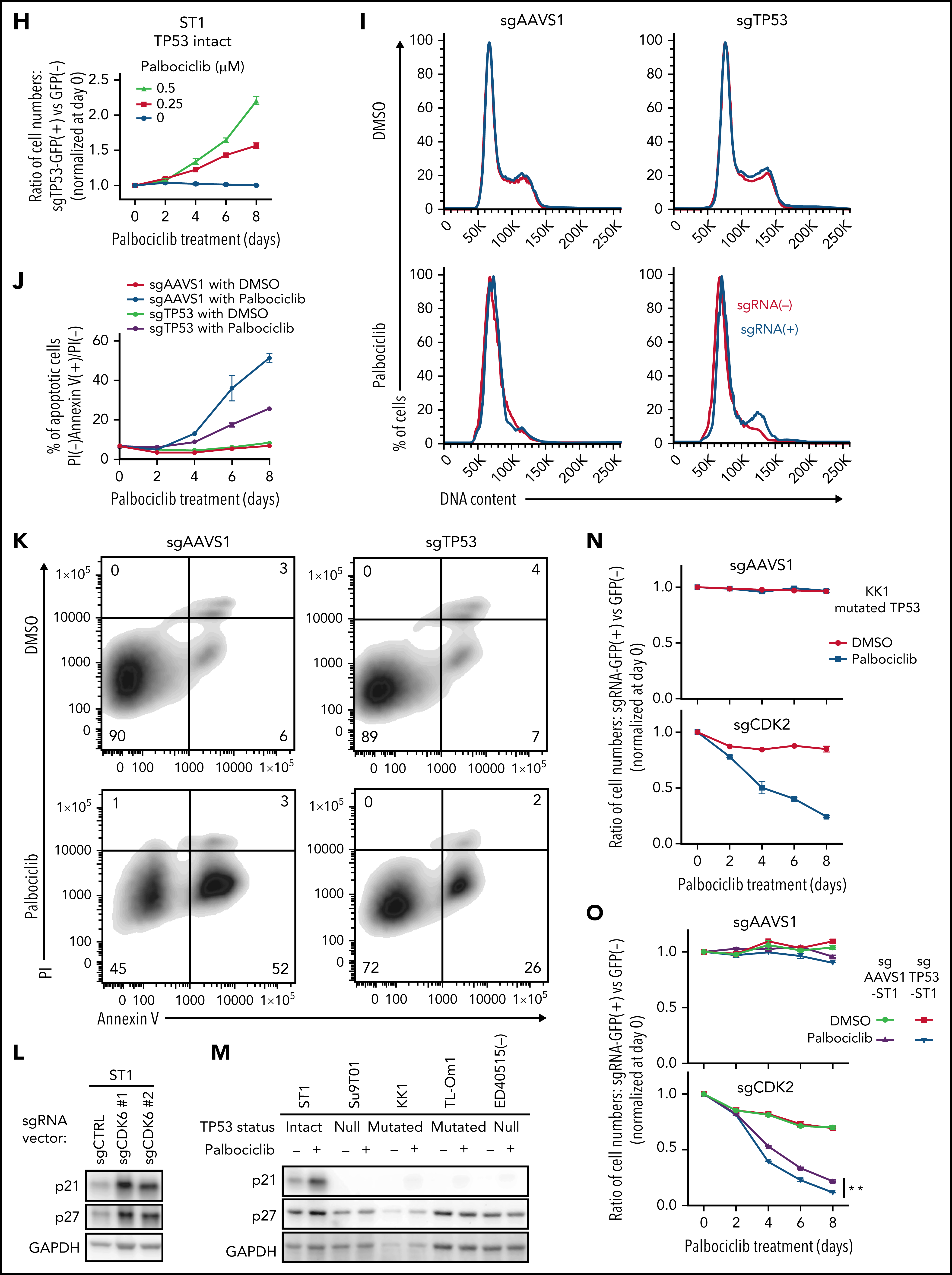
通过对CRISPR筛选结果进行通路分析,发现ATLL细胞依赖mTORC1信号通路。MTOR敲除能够抑制多种ATLL细胞生长,免疫印迹显示ST1细胞中mTOR下游通路分子处于磷酸化状态。鉴于既往研究显示CDK和mTOR抑制剂联合用药对不同类型肿瘤的有益效果,作者利用mTOR抑制剂与帕博西尼处理不同ATLL细胞系,结果显示mTOR抑制剂与帕博西尼可以协同抑制不同TP53状态的ATLL细胞,两抑制剂联合作用对Rb磷酸化水平的抑制效果也高于分别单独用药。
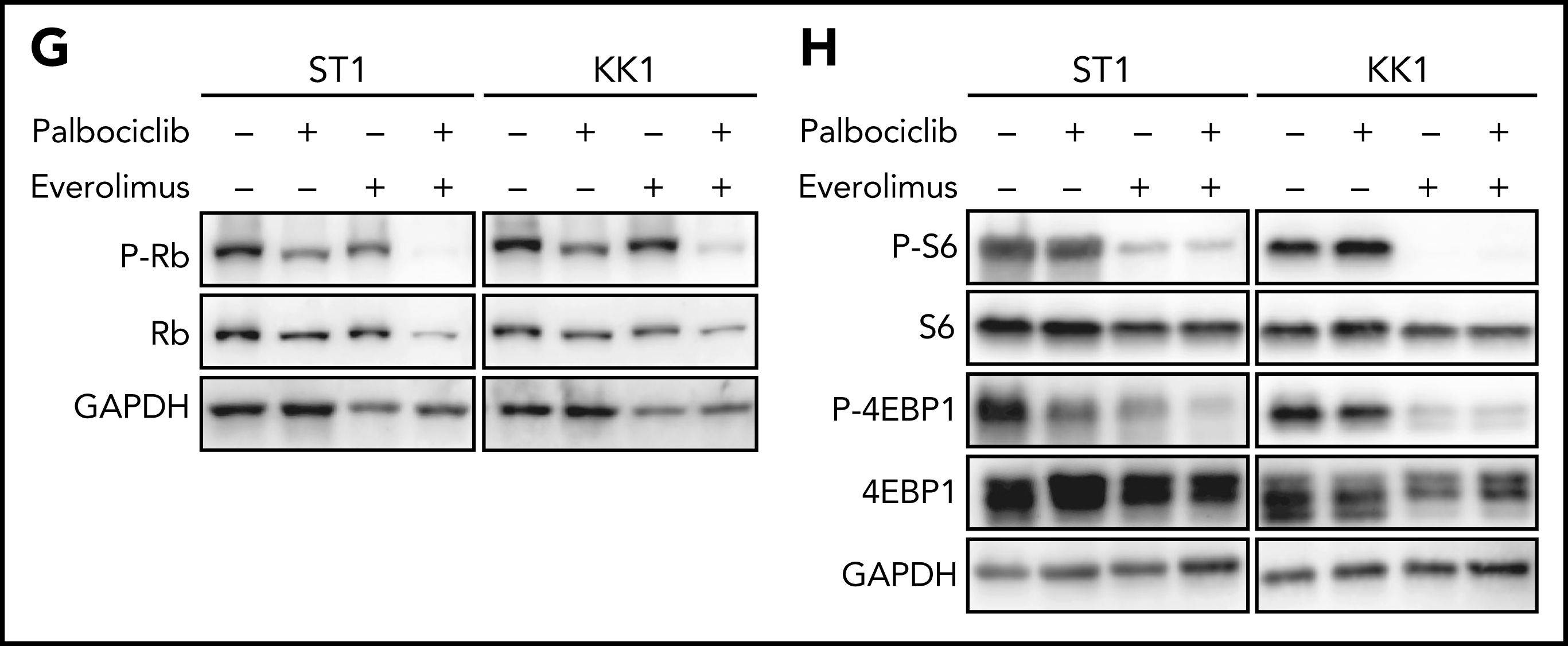
作者进一步对ATLL患者原代细胞进行探究,与细胞系实验结果一致,帕博西尼能够浓度依赖地抑制ATLL原代细胞增殖,在联合mTOR抑制剂后抑制程度增加,而对其他正常的T细胞没有明显影响。为在体内水平探究联合用药的安全性和有效性,作者将ATLL细胞接种至小鼠体内建立异种移植模型并给药,结果显示mTOR抑制剂与帕博西尼分别单独用药时即可抑制移植瘤生长,联合用药后的抑制效果则更加显著,同时药物处理对小鼠体重没有显著影响,证明这一策略的安全性和有效性。

扫一扫,反馈当前页面
和元生物
