

【Circulation Research】宝贝计划,宝宝的小心脏有HDAC3来守护
前言
距离全球首例猪心脏移植手术已经过去半年,“顶流”马里兰大学医学院Dr. Deqiang Li的研究团队于2022年7月在Circulation Research杂志上发表题目为“Epicardial HDAC3 promotes myocardial growth through a novel microRNA pathway”的心血管文章。该研究揭示了心脏发育中心外膜HDAC3通过抑制miR-322/miR-503来刺激Fgf9和Igf2进而促进致密心肌生长,为阐明先天性心脏缺陷的病因和促进心肌再生提供了新的策略和见解。
背景介绍
HDAC(组蛋白去乙酰化酶)3是一类HDAC家族的成员,它可以催化从组蛋白尾部赖氨酸残基中去除乙酰基,并通过调节基因表达参与许多生物学过程。中胚层或全基因敲除HDAC3可导致肌源性缺陷和早期胚胎致死,心肌中特异性的HDAC3缺失却不会在早期心脏发育中对心脏形态表型产生影响,而是会损害出生后后期的心脏功能。这表明HDAC3在非肌细胞室间的功能可能对早期心肌发育至关重要。本研究旨在探寻早期心脏发育过程中,HDAC3在心外膜中的作用以及Fgf9和Igf2的表达如何受到HDACs或miRs的调控。
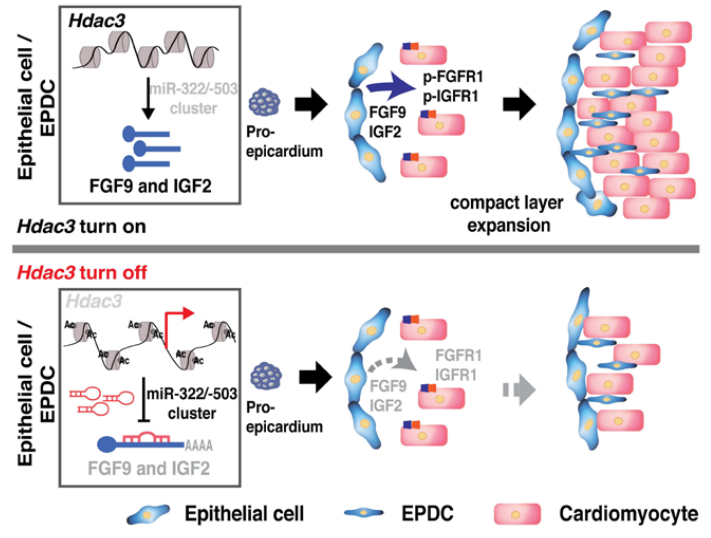
图. The schematics of the working model
亮点要素
1、发育中的心外膜中Hdac3的表达对致密心肌生长至关重要
2、HDAC3对于心外膜衍生的细胞分化和迁移是很重要的
3、miR-322和miR-503抑制Fgf9和Igf2的转录
4、HDAC3通过抑制miR-322和miR-503的表达来促进Fgf9和Igf2的转录
研究内容
01
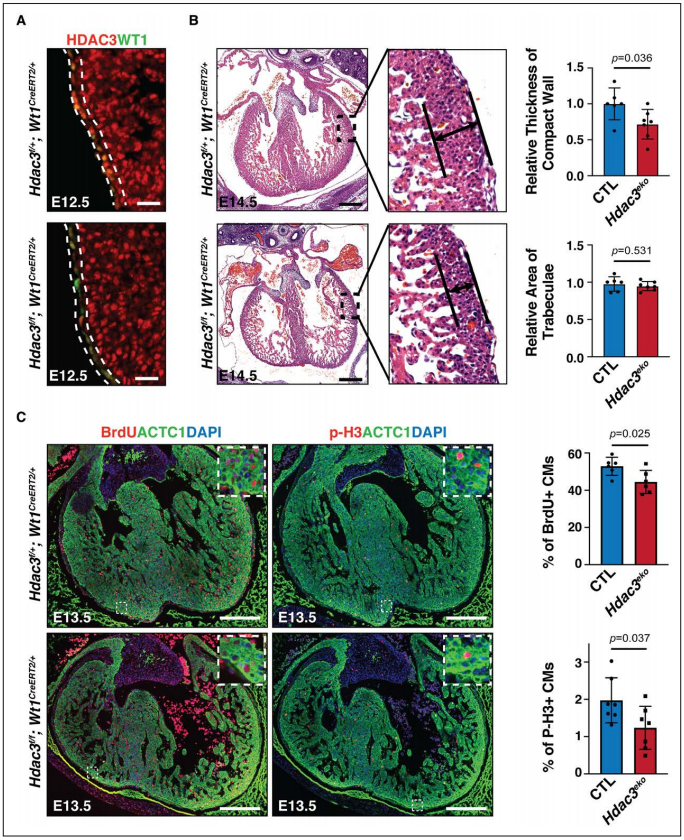
Figure 1. Epicardial deletion of Hdac3 resulted in hypoplasia of ventricular compact wall.
02
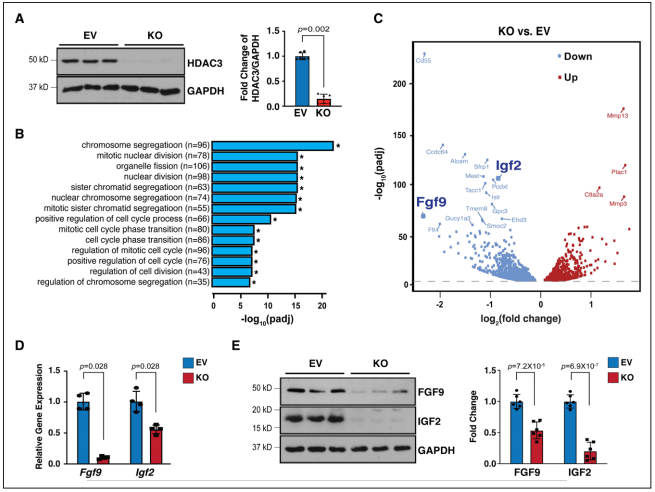
Figure 2. Hdac3 deletion resulted in downregulation of Fgf9 and Igf2.
03
研究者试图确定Fgf9和Igf2的减少是否与心肌细胞增殖减少有关。与对照细胞EV相比,Hdac3敲除株KO上清液中Fgf9和Igf2显著降低。又用细胞上清处理E13.5分离的原代胚胎心肌细胞。与EV上清相比,KO上清处理的pH3阳性心肌细胞百分比和心肌细胞总数显著降低并且其减弱了心肌细胞增殖下游信号通路ERK磷酸化的激活。而在KO中补充Fgf9和Igf2可以修复心肌细胞增殖缺陷并使p-FGFR1/p-IGFR1活化。以上结果表明,来自心外膜细胞分泌的Fgf9和Igf2提供了驱动心肌细胞增殖的重要线索。
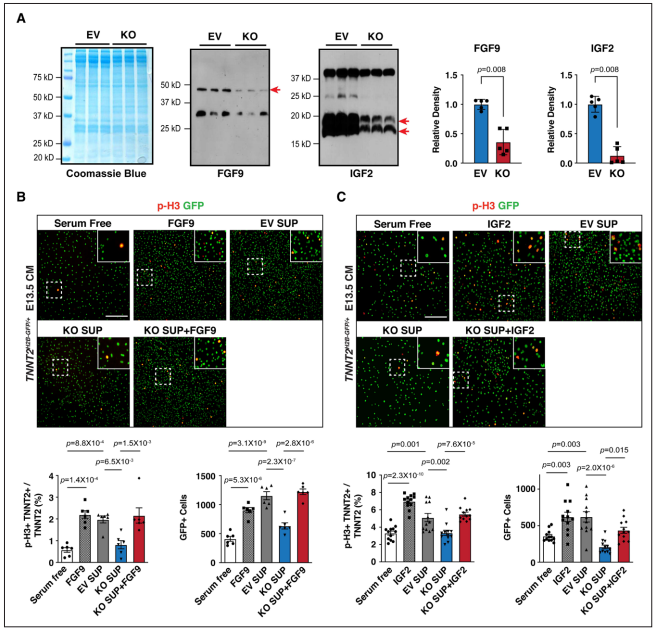
Figure 3. Supplementation of Fgf9 or Igf2 rescues CM proliferation defects.
04
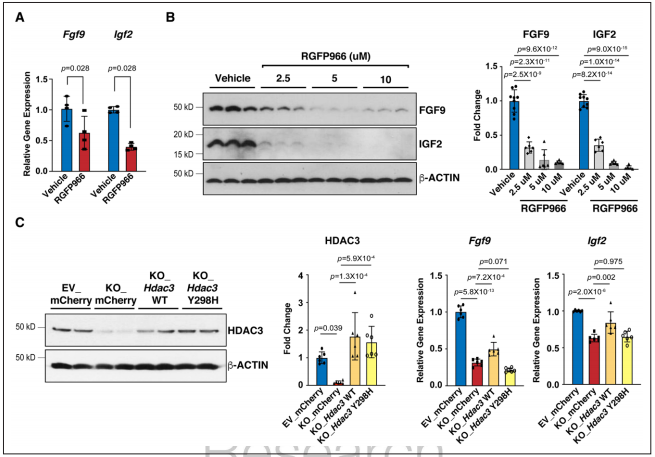
Figure 4. HDAC3 induces the expression of Fgf9 and Igf2 dependent on its deacetylase activity.
05
Fgfs和Igfs的表达可被miRs调控,研究者对敲除株进行了miR测序。通过差异表达分析及模拟结合位点筛选发现miR-322或miR-503模拟物处理显著抑制Fgf9和Igf2的表达。通过Western blot进一步验证了miR-322 mimics/miR-503 mimics处理对Fgf9和Igf2表达的显著抑制作用。又使用miR-322或miR-503模拟物处理培养的E13.5心肌细胞,显著降低了pH3阳性心肌细胞的百分比,表明miR-322和miR-503抑制心肌细胞增殖。当HDAC3的去乙酰化酶活性被RGFP966抑制时,miR-322和miR-503与敲除株中同样显著上调。体内E13.5 eko心脏中也观察到miR-322和miR-503的显著上调。证明了miR-322、miR-503能够抑制Fgf9、Igf2表达和心肌细胞增殖。
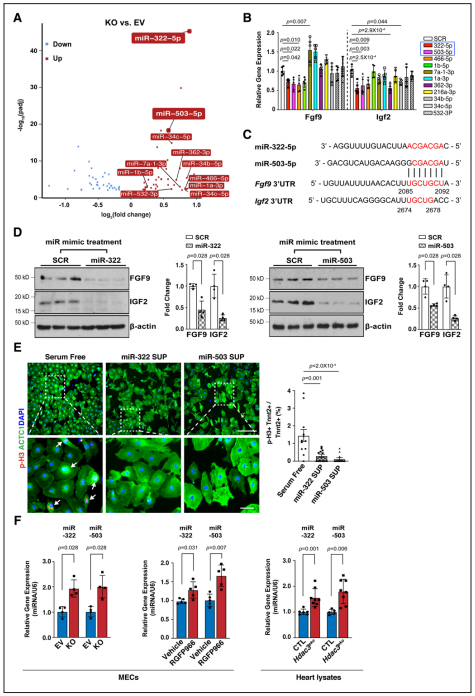
Figure 5.miR-322 and miR-503 repress the expression of Fgf9 and Igf2 and CM proliferation.
06

Figure 6. Knockdown of miR-322 or miR-503 restores the expression of Fgf9 and Igf2.
07
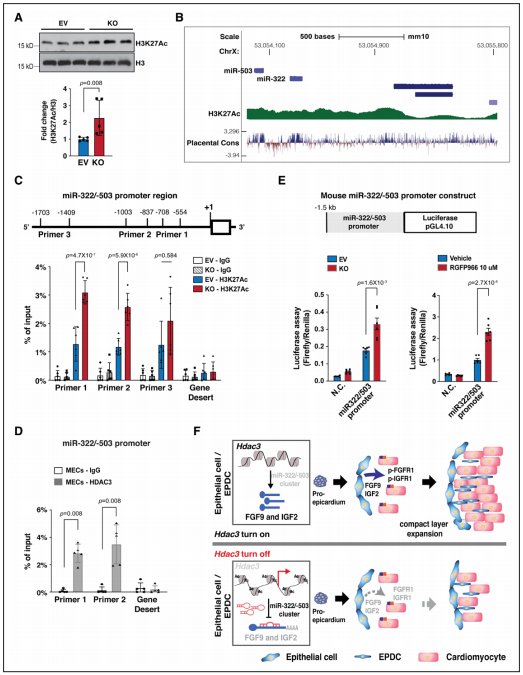
Figure 7. HDAC3 represses miR-322/miR-503 promoter activity.
解暑好物!心血管及代谢疾病动物模型整体实验8折优惠!夏日解忧实验室!
和元生物具有多年丰富的动物实验和心血管动物模型构建的实操经验,并兼具出色的基因操作与病毒构建等技术。同时依托公司的检测平台、病理平台、细胞平台等专业实验平台,可为您提供从心血管动物模型到基因干预、病理检测、细胞功能学研究等一站式心血管研究服务体系,您可体验文中涉及到的心血管前沿研究手段与支持。欢迎关注和元公众号了解实验服务详情,和元生物期待您的到来。

扫一扫,反馈当前页面
和元生物
