

【综述】一文全面了解肌营养不良症及多款基因疗法
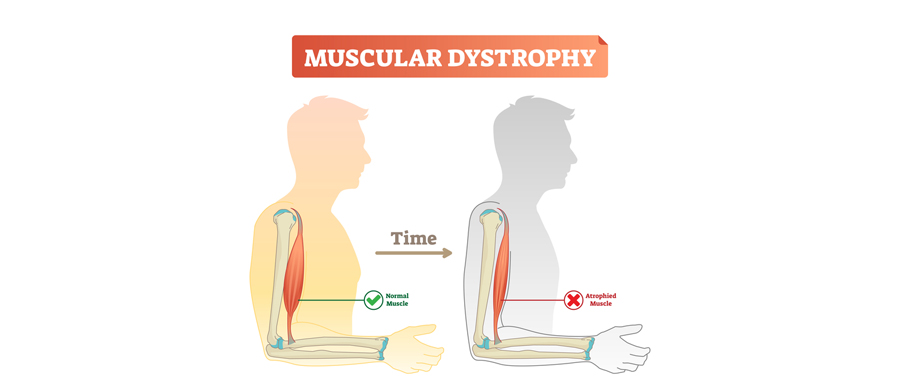
肌营养不良症(Muscular dystrophy,MD)是一组遗传性疾病,可导致患者发生进行性肌无力和肌丧失。目前已知的MD类型超过30多种,其病因和症状各不相同。最常见的MD类型出现在儿童时期,也有少量MD只影响成年人。
MD通常是由关键的、合成肌肉所需蛋白的基因突变引起的,患者通常从父母那里遗传这些突变,但是也有少量MD可以从新的基因突变中产生。MD难以治愈,但现有的治疗方法可以减轻症状,减缓疾病的发展。本文将介绍MD的基本类型以及针对MD开发的多款前沿基因治疗疗法,为广大基因治疗从业者分享信息。
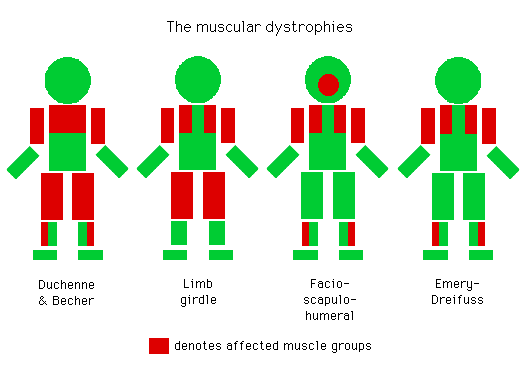
提到肌营养不良症,或肌肉萎缩症,我们首先想到的是杜氏肌营养不良(DMD),但除了DMD,还有30多种肌营养不良类型,可大致分为常见类型的肌营养不良症和罕见的先天肌营养不良症,不同肌营养不良症影响的肌肉区域不同。
TMD是一种以踝关节肌肉无力和消瘦为特征的肌营养不良症,该疾病可进展到小腿前部胫骨(胫骨)的肌肉,该类型TMD在芬兰很普遍。
先天性肌营养不良(Congenital muscular dystrophies (CMDs))指的是一系列肌营养不良症,其特征是从出生或生命早期开始就出现肌肉无力和消瘦(萎缩)。
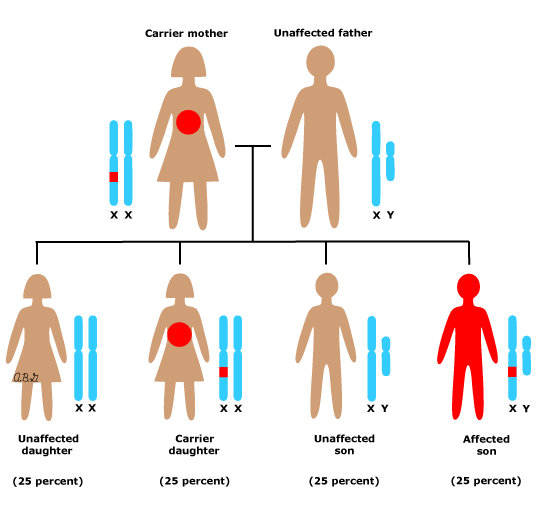
CMDs同样是由基因突变引起的,可能是遗传的,也可能是再生的。主要包括:
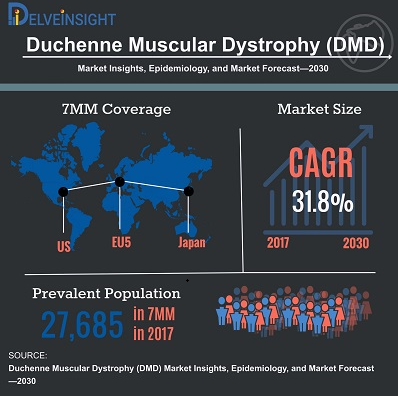
目前,针对肌营养不良症的大量基因疗法正在研发中,人们试图通过修正基因达到对肌营养不良症的痊愈。根据资本市场的评估,以研究最为火热的DMD为例,未来10年的DMD药物年复合增长率(CAGR)达到31.8%,有巨大的发展空间。
针对MD的基因疗法主要包括基于AAV的基因治疗、外显子跳跃、RNA干扰等。
腺相关病毒(Adeno-associated virus, AAVs)是基因治疗常用的病毒载体之一,这是一种人工改造的工具病毒载体,其作用是将功能基因直接运送到特定组织的细胞中。目前,基于AAV载体的基因治疗MD主要包括如下药物:

PF-06939926疗法借助AAV9将微小肌营养不良蛋白基因(DMD基因的一个较短但具有功能的副本)递送给肌肉组织,在其中表达短的Dystrophin蛋白,从而一次性减缓或阻止DMD患者的肌肉退化。
2016年,辉瑞收购Bamboo Therapeutics,从而获得Bamboo旗下针对DMD的基因疗法PF-06939926。PF-06939926于2017年5月获得美国食品和药物管理局(FDA)和欧洲药品管理局(EMA)的孤儿药认定,并获得FDA罕见儿科疾病认定。

针对PF-06939926的Ib期临床试验是在美国开展的多中心试验,用于评估15位患有DMD的男孩对该药物的安全性、耐受性和初步有效性。目前,该试验仍在招募年龄在4至12岁的受试患者。受试者按1*10^14vg/Kg体重(低剂量组)或3*10^14vg/Kg体重(高剂量组)一次性静脉输注PF-06939926,治疗后随访5年。该试验的主要目标是评估PF-06939926的安全性和耐受性,次要目标包括评估肌肉活检中微小肌营养不良蛋白的水平和定位。此外,评价运动功能的NorthStar Ambulatory Assessment (NSAA)等级量表情况的变化,以及肌肉脂肪分数(DMD严重程度的标志)的变化亦是该试验的检验目标。
PF-06939926有潜力成为第一个进入III期临床试验的DMD基因治疗药物,并开始使用商业规模的病毒生产。而现阶段的大规模生产能力将有助于Pfizer在药物被批准上市后迅速让患者受惠。

rhLAM -111(重组人laminin-111)是Prothelia开发的一种实验性基因疗法,目前正在美国、加拿大和欧盟的实验室进行临床前研究,可用于治疗DMD和先天性1A型肌营养不良症(MDC1A)。
在MDC1A患者中,rhLAM -111可以在疾病早期取代层粘连蛋白,逐步消除肌营养不良的症状。rhLAM -111还能增加α7β1整合素和相关蛋白的产生。α7β1整合素是另一种结构蛋白,该疗法希望增加这些蛋白防止肌营养不良带来的流失。此外,rhLAM -111被认为可以刺激肌肉愈合途径。
研究人员正在开发一种基因疗法,可以增加肌肉组织中rhLAM -111的产量。这种疗法将包含一种工具载体病毒,编码rhLAM -111的基因。人们希望感染病毒的细胞开始产生rhLAM -111,从而增加α7β1整合素和utrophin的产生,并刺激肌肉损伤的修复。
该方法的第一阶段临床试验预计将于2021年中期开始。

Solid旗下的SGT-001是一种新型的腺相关病毒(AAV)介导的基因疗法,旨在解决杜氏肌营养不良症的潜在基因问题。DMD是由Dystrophin基因突变引起的,其突变导致Dystrophin蛋白缺失或接近缺失。
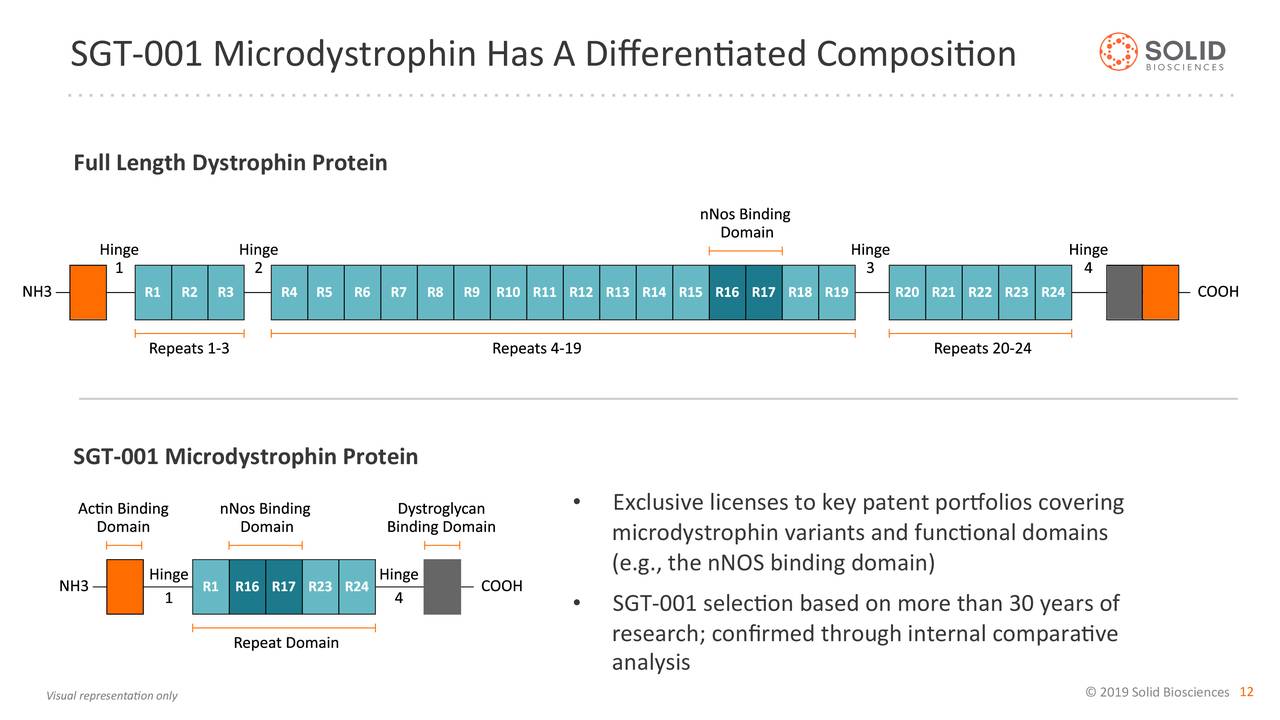
SGT-001是系统给药、治疗DMD的候选基因治疗药物,负责将外源Microdystrophin基因传递到体内。该基因编码一种在肌肉中表达的功能性蛋白替代物,并稳定必要的相关蛋白,包括神经元一氧化氮合酶(nNOS)。Solid临床前试验数据表明,无论基因突变或疾病阶段如何,SGT-001都有可能减缓或停止Duchenne的进展。目前,SGT-001已被授予美国和欧盟的孤儿药物认定。
高能预警,下列4个疗法均由Sarepta Therapeutics开发,其针对各种基因遗传疾病的基因疗法管线多达42条。
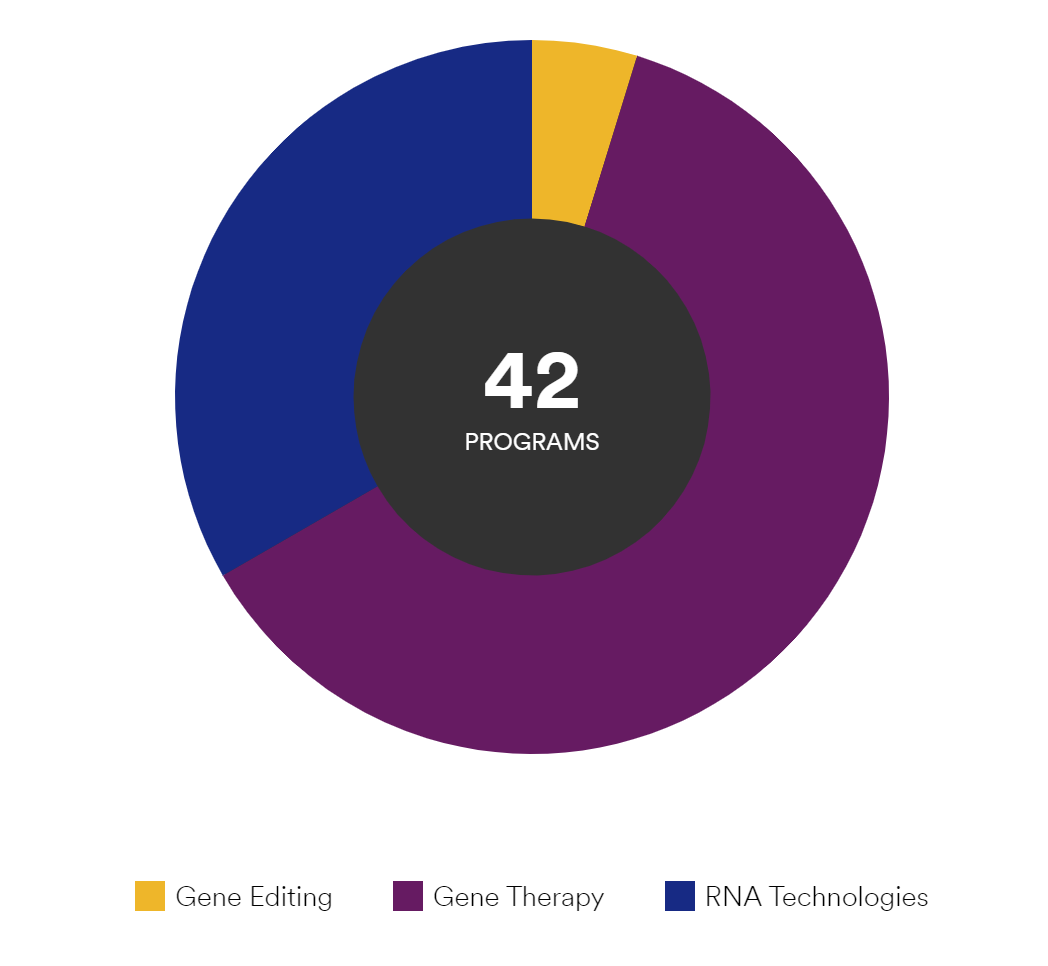

GALGT2 (B4GALNT2)是由Sarepta Therapeutics公司开发的治疗杜氏肌营养不良症(DMD)的实验性基因疗法,该疗法与美国全国儿童医院合作进行临床试验。
GALGT2基因疗法借助人工改造的rAAVrh74腺相关病毒载体将GALGT2基因导入体内,并在骨骼肌和心肌细胞中表达GALGT2基因。GALGT2基因则可以上调或增加其他对肌肉功能至关重要的蛋白质表达量,从而逐步修复肌肉功能。由于GALGT2不能取代有缺陷的DMD基因,故该疗法被称为替代基因疗法。该疗法不仅有可能纠正由DMD基因突变引起的肌功能异常,还有可能纠正其他肌营养不良症,如肢带型肌营养不良2A (LGMD2A)和1A型先天肌营养不良(MDC1A)。
目前,GALGT2基因疗法已经进入临床I/II期、开放标签试验(NCT03333590),用于评估该疗法的安全性和有效性。试验中,6名DMD患者接受低剂量或高剂量的GALGT2基因治疗,治疗后的24个月内定期对他们进行评估。该试验结果预计将于2021年11月公布。

SRP-9001(微营养不良蛋白基因治疗)疗法是治疗DMD的候选基因,该疗法由全国儿童医院的科学家研制开发,并授权给Sarepta Therapeutics公司,完成SRP-9001的临床推进。
SRP-9001疗法被开发在患者体内表达小版本的、携带特定的功能区域的肌营养不良蛋白Microdystrophin。微营养不良蛋白的工作原理与正常的营养不良蛋白相似,只是尺寸更小。该疗法同样选取rAAVrh74腺相关病毒载体作为基因递送工具,同时采用肌肉特异性启动子MHCK7,增强心脏和骨骼肌中的基因表达。
目前,SRP-9001的II期临床试验(NCT03769116)招募40名年龄在4到7岁之间的DMD患者,以评估注射SRP-9001的安全性和有效性。试验将采用随机、双盲和安慰剂对照。研究结果预计将于2022年10月公布。

SRP-9003(原MYO-101和scAAVrh74.MHCK7.hSGCB)是治疗肢体束带型肌营养不良(LGMD) 2E型的候选基因治疗疗法。
2E型LGMD(LGMD2E)是一种由SGCB基因突变引起的肌营养不良症,SGCB基因的作用是生成β-sarcoglycan蛋白,β-sarcoglycan是一种更大的蛋白质sarcoglycan protein complex的一部分,这种蛋白质对肌肉组织的健康至关重要,因而LGMD患者的肌组织缺乏β-sarcoglycan。SRP-9003借助scAAVrh74和MHCK7启动子,向骨骼、心脏和横膈膜肌肉传递人类SGCB基因的功能性副本,来恢复肌肉组织中的β-sarcoglycan水平。
SRP-9003于2018年4月获得美国食品和药物管理局(FDA)的孤儿药指定。SRP-9003开发项目也被FDA授予罕见小儿疾病的称号,这使得该疗法有资格获得优先审查,可能会加速批准。

SRP-9004 (MYO-102)是Sarepta Therapeutics公司开发的一种实验性基因疗法,用于治疗肢体束带型肌营养不良症2D (LGMD2D),这是肢体束带型肌营养不良症的一种亚型,常在儿童早期被诊断。
LGMD2D是由SGCA基因突变引起的,SGCA基因用于编码α-sarcoglycan蛋白(sarcoglycan蛋白的一个亚基),sarcoglycan通常位于肌细胞周围的细胞膜上,通过结合和稳定肌营养不良蛋白复合物来帮助维持肌肉组织的结构。
SRP-9004由AAV携带正常的SGCA基因副本,在细胞内产生α-sarcoglycan蛋白,从而允许sarcoglycan正常组装并与肌营养不良蛋白复合物相互作用,纠正患者SGCA基因突变引起的问题。SRP-9004于2019年1月被美国食品和药物管理局(FDA)指定为孤儿药。
除了上述4款针对MD的药物,Sarepta Therapeutics更多的基因治疗研发管线还包括:
外显子是基因表达的关键部分,编码表达蛋白的基因序列。外显子跳跃(Exon skipping)则是借助反义寡核苷酸(AO),对基因缺失或外显子突变部分的“修补”。通过外显子跳跃,细胞会产生一种截短的但具有功能的蛋白,以缓解肌营养不良症表征。
由于DMD和BMD均是在DMD基因的几个位置上缺失外显子,因而外显子跳跃目前针对该两种疾病开展试验。
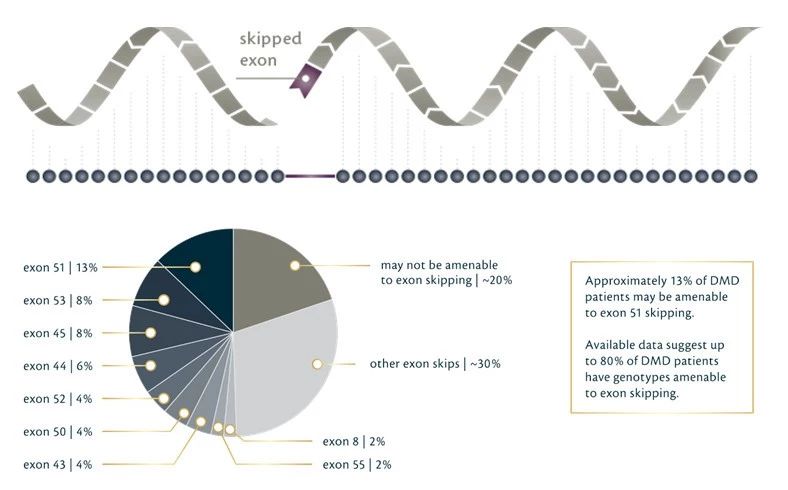
Exondys 51 (eteplirsen)是FDA批准的一种治疗方法,来自Sarepta,该疗法在DMD患者中跳过exon51,缓解DMD症状。
其他正在研究的外显子跳跃的疗法的包括同样来自Sarepta的跳过外显子53的Golodirsen (SRP-4053),跳过外显子45的SRP-4045‘以及结合到突变位点以便正确读取基因的DS-5141(来自Daiichi Sankyo)等。

来自PTC Therapeutics的创新疗法Translarna正在对DMD和BMD患者开展临床研究,以逆转无意义突变带来的患者体内肌营养不良蛋白表达下降的现象。Translarna在欧洲、以色列和韩国的大部分地区被批准用于治疗无意义突变的DMD,但在美国未获批准。
庆大霉素是一种抗生素,用于治疗细菌感染,然而近期一项研究发现庆大霉素在提高肌营养不良蛋白水平和降低血清肌酸激酶水平方面是有效的。

RNA干扰(RNAi)是抑制或阻断信使RNA (mRNA)功能的过程。信使RNA包含了来自基因的遗传信息,用于合成蛋白质。
来自Benitec Biopharma的候选药物BB-301是一种实验性的RNAi疗法,目前正在研究用于治疗眼咽部肌营养不良症(OPMD)。BB-301利用RNAi阻止突变的PABPN1基因通过mRNA产生异常的PABPN1蛋白。同时,它将一个健康的PABPN1基因拷贝导入细胞,做到替代型基因疗法。
除了本文介绍的基因疗法,还有大量的针对MD的基因疗法处于研发的不同阶段,大量制药公司与研究机构开展的基因治疗疗法开发合作也为该领域的突破带来帮助,如Santhera,未来,Obio GeneTherapy将持续关注各种疾病领域的基因治疗疗法开发,也希望基因治疗能够早日落地,真正造福人类。
作为一家新型基因和细胞治疗产业化CDMO服务公司,和元生物 可提供非注册临床研究用质粒和病毒生产、基因治疗新药临床申报整体方案、基因治疗临床样品及商业化GMP生产等服务,涉及产品包括基因和细胞治疗用质粒、慢病毒、腺相关病毒、溶瘤病毒、新型基因疫苗等,同时欢迎基于外泌体的药物开发合作。
以“基因药·中国造”为使命,助力基因治疗造福人类的伟大愿景。


扫一扫,反馈当前页面
和元生物
